A collaborative effort between research groups at the High Energy Accelerator Research Organization (KEK), Tsukuba Japan and the University of Hawaii has obtained results that pushed the frontiers of fundamental science and provide a key breakthrough for the development of practical hydrogen powered vehicles. The paper reporting these results obtained by the KEK group led by Prof. Ryosuke Kadono, Dr. Etsuo Akiba of AIST, and the UH group led by Prof. Craig M. Jensen has been accepted by the prestigious publication, Physical Review Letters became available on the web on January 15.
A major obstacle to the conversion of the world to a "hydrogen economy" is the problem of onboard hydrogen storage. High-pressure and cryogenic hydrogen-storage systems are impractical for vehicular applications due to safety concerns and volumetric constraints. This has prompted an extensive effort to develop solid hydrogen-storage systems for vehicular application. Metal hydrides, advanced carbons, metal organic frameworks have been investigated as hydrogen carriers. Unfortunately, despite decades of extensive effort no material has been found which has the combination of a high gravimetric hydrogen density, adequate hydrogen-dissociation energetics, reliability, and low cost required for commercial vehicular application. Group I and II salts of [AlH4]-, [NH2]-, and [BH4]- (alanates, amides, and borohydrides) have recently received considerable attention as potential hydrogen storage materials and are currently referred to as "complex hydrides". These materials have high hydrogen gravimetric densities and are, in most cases, commercially available. Thus a priori, they would seem to be viable candidates for application as practical, on-board hydrogen-storage materials. Many of these "complex hydrides" have, in fact, been utilized in "one-pass" hydrogen-storage systems in which hydrogen is evolved from the hydride upon contact with water. However, the hydrolysis reactions are highly irreversible and could not serve as the basis for rechargeable hydrogen-storage systems. The thermodynamics of the direct, reversible dehydrogenation of some complex hydrides lie within the limits that are required for a practical, onboard hydrogen carrier. All of these materials are, however, plagued by high kinetics barriers to dehydrogenation and/or re-hydrogenation in the solid-state. Traditionally, it was thought that it would be impossible to reduce the barrier heights to an extent that would give reaction rates that even approached those that would be required for vehicular applications. This situation was changed about 10 years ago by the pioneering work at the Max Planck Institute in Germany that demonstrated that upon doping with selected titanium compounds, the dehydriding of anionic aluminum hydrides could be kinetically enhanced and rendered reversible under moderate conditions in the solid state. This breakthrough has led to a world-wide effort to develop complex hydrides as hydrogen storage materials. An overview of these research and development activities appear in an article co-authored by Prof. Jensen in the special thematic "Hydrogen" issue of Chemical Reviews this past October. His group at the University of Hawaii has long been a leader in this effort and Jensen was awarded "2004 R&D Award" by the U. S. Department of Energy Hydrogen Program for these activities.
In the decade that has passed since Ti-doping was found to accelerate kinetics of the reversible dehydrogenation of these materials, a clear understanding on the microscopic mechanism action has been slow to emerge. This is because it involves solid state effects that are unlike anything that was previously observed. The lack of fundamental understanding of what the dopants are doing in these materials has been a roadblock to advancing complex hydrides to the point of practical viability. Thus gaining an understanding of this phenomenon not only pushs the frontiers of physical science but is also of great value to the future choice/design of complex hydrides to be utilized as practical hydrogen storage materials. When the UH team noted that the effect seemed to involve a very small amount of Ti generated defects in the material, they enlisted the aid Prof. Kadono and his team at KEK to help solve the problem because they are one of the world's leading groups in using to muons to probe defects that can not be characterized any other way. Using materials synthesized at the University of Hawaii and the muon expertise at KEK, the origin of slow dehydrogenation kinetics in a complex hydride-based hydrogen storage material and the mechanism of accelerating it by doping with transition metal catalysts has been revealed for the first time by the novel technique of muon spin rotation. This work shows that interstitial hydrogen atoms in sodium alanate, a prototype complex hydride are trapped a bonding interaction with negatively charged alanate ions. This interaction is a new variation in hydrogen bonding that has not been previously considered in hydrogen storage materials. The studies indicate that the Ti acts to reduce the kinetic barrier for hydrogen to move from the hydrogen bonding traps to the octahedral interstitial site where H atoms are mobile, thus accelerating the H-kinetics.
| [ Related Web Site ] |
Muon Science Laboratory |
| [ Media Contact ] |
Ryosuke Kadono, KEK Insititue of Materials Structure Science
+81 29-864-5625
Youhei Morita, KEK Public Relations Office
+81 29-879-6047
|
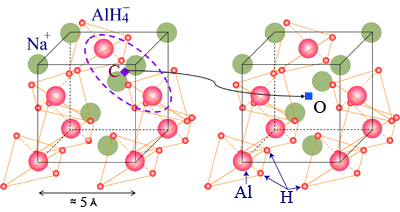 |
| Figure 1 : Crystal structure of sodium alanate. A muon (equivalent of hydrogen) forms a complex state with two nearest neighboring alanate ions (shown by tetrahedra) at the C site. Adding titanium (not shown here) reduces the energy required for muons (and H atoms) to move from C site to another interstitial O site, where muons (and H atoms) undergo longrange diffusive motion. |
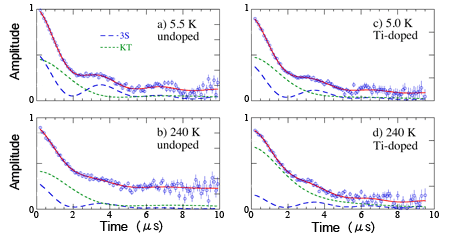 |
| Figure 2 : Signals form muons implanted into sodium alanate, where long-dashed curves (denoted as 3S) are muons at the C site that exhibit a characteristic oscillation with time. Those of short-dashed curves represent signals from muons sitting at the O site (denoted as KT). Adding titanium enhances the degree of conversion from C site to O site upon elevating temperature (c-d) than that in the undoped case (a-b). |
|

